Neuroscience

Abstract
Munc13 proteins are essential regulators of neurotransmitter release at nerve cell synapses. They mediate the priming step that renders synaptic vesicles fusion-competent, and their genetic elimination causes a complete block of synaptic transmission. Here we have described a patient displaying a disorder characterized by a dyskinetic movement disorder, developmental delay, and autism. Using whole-exome sequencing, we have shown that this condition is associated with a rare, de novo Pro814Leu variant in the major human Munc13 paralog UNC13A (also known as Munc13-1). Electrophysiological studies in murine neuronal cultures and functional analyses in
Authors
Noa Lipstein, Nanda M. Verhoeven-Duif, Francesco E. Michelassi, Nathaniel Calloway, Peter M. van Hasselt, Katarzyna Pienkowska, Gijs van Haaften, Mieke M. van Haelst, Ron van Empelen, Inge Cuppen, Heleen C. van Teeseling, Annemieke M.V. Evelein, Jacob A. Vorstman, Sven Thoms, Olaf Jahn, Karen J. Duran, Glen R. Monroe, Timothy A. Ryan, Holger Taschenberger, Jeremy S. Dittman, Jeong-Seop Rhee, Gepke Visser, Judith J. Jans, Nils Brose

The chromatin remodeling factor CHD7 controls cerebellar development by regulating reelin expression
Abstract
The mechanisms underlying the neurodevelopmental deficits associated with CHARGE syndrome, which include cerebellar hypoplasia, developmental delay, coordination problems, and autistic features, have not been identified. CHARGE syndrome has been associated with mutations in the gene encoding the ATP-dependent chromatin remodeler CHD7. CHD7 is expressed in neural stem and progenitor cells, but its role in neurogenesis during brain development remains unknown. Here we have shown that deletion of
Authors
Danielle E. Whittaker, Kimberley L.H. Riegman, Sahrunizam Kasah, Conor Mohan, Tian Yu, Blanca Pijuan Sala, Husam Hebaishi, Angela Caruso, Ana Claudia Marques, Caterina Michetti, María Eugenia Sanz Smachetti, Apar Shah, Mara Sabbioni, Omer Kulhanci, Wee-Wei Tee, Danny Reinberg, Maria Luisa Scattoni, Holger Volk, Imelda McGonnell, Fiona C. Wardle, Cathy Fernandes, M. Albert Basson
Abstract
Authors
Alexander N. Comninos, Matthew B. Wall, Lysia Demetriou, Amar J. Shah, Sophie A. Clarke, Shakunthala Narayanaswamy, Alexander Nesbitt, Chioma Izzi-Engbeaya, Julia K. Prague, Ali Abbara, Risheka Ratnasabapathy, Victoria Salem, Gurjinder M. Nijher, Channa N. Jayasena, Mark Tanner, Paul Bassett, Amrish Mehta, Eugenii A. Rabiner, Christoph Hönigsperger, Meire Ribeiro Silva, Ole Kristian Brandtzaeg, Elsa Lundanes, Steven Ray Wilson, Rachel C. Brown, Sarah A. Thomas, Stephen R. Bloom, Waljit S. Dhillo

Abstract
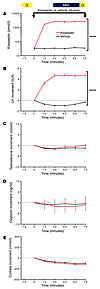
Olfactory dysfunction is broadly associated with neurodevelopmental and neurodegenerative diseases and predicts increased mortality rates in healthy individuals. Conventional measurements of olfactory health assess odor processing pathways within the brain and provide a limited understanding of primary odor detection. Quantification of the olfactory sensory neurons (OSNs), which detect odors within the nasal cavity, would provide insight into the etiology of olfactory dysfunction associated with disease and mortality. Notably, OSNs are continually replenished by adult neurogenesis in mammals, including humans, so OSN measurements are primed to provide specialized insights into neurological disease. Here, we have evaluated a PET radiotracer, [11C]GV1-57, that specifically binds mature OSNs and quantifies the mature OSN population in vivo. [11C]GV1-57 monitored native OSN population dynamics in rodents, detecting OSN generation during postnatal development and aging-associated neurodegeneration. [11C]GV1-57 additionally measured rates of neuron regeneration after acute injury and early-stage OSN deficits in a rodent tauopathy model of neurodegenerative disease. Preliminary assessment in nonhuman primates suggested maintained uptake and saturable binding of [18F]GV1-57 in primate nasal epithelium, supporting its translational potential. Future applications for GV1-57 include monitoring additional diseases or conditions associated with olfactory dysregulation, including cognitive decline, as well as monitoring effects of neuroregenerative or neuroprotective therapeutics.
Authors
Genevieve C. Van de Bittner, Misha M. Riley, Luxiang Cao, Janina Ehses, Scott P. Herrick, Emily L. Ricq, Hsiao-Ying Wey, Michael J. O’Neill, Zeshan Ahmed, Tracey K. Murray, Jaclyn E. Smith, Changning Wang, Frederick A. Schroeder, Mark W. Albers, Jacob M. Hooker

Abstract
Parkinson’s disease (PD) patients experience loss of normal motor function (hypokinesia), but can develop uncontrollable movements known as dyskinesia upon treatment with L-DOPA. Poverty or excess of movement in PD has been attributed to overactivity of striatal projection neurons forming either the indirect (iSPNs) or the direct (dSPNs) pathway, respectively. Here, we investigated the two pathways’ contribution to different motor features using SPN type–specific chemogenetic stimulation in rodent models of PD (PD mice) and L-DOPA–induced dyskinesia (LID mice). Using the activatory Gq-coupled human M3 muscarinic receptor (hM3Dq), we found that chemogenetic stimulation of dSPNs mimicked, while stimulation of iSPNs abolished the therapeutic action of L-DOPA in PD mice. In LID mice, hM3Dq stimulation of dSPNs exacerbated dyskinetic responses to L-DOPA, while stimulation of iSPNs inhibited these responses. In the absence of L-DOPA, only chemogenetic stimulation of dSPNs mediated through the Gs-coupled modified rat muscarinic M3 receptor (rM3Ds) induced appreciable dyskinesia in PD mice. Combining D2 receptor agonist treatment with rM3Ds-dSPN stimulation reproduced all symptoms of LID. These results demonstrate that dSPNs and iSPNs oppositely modulate both therapeutic and dyskinetic responses to dopamine replacement therapy in PD. We also show that chemogenetic stimulation of different signaling pathways in dSPNs leads to markedly different motor outcomes. Our findings have important implications for the design of effective antiparkinsonian and antidyskinetic drug therapies.
Authors
Cristina Alcacer, Laura Andreoli, Irene Sebastianutto, Johan Jakobsson, Tim Fieblinger, Maria Angela Cenci

Abstract
Sensory neurons have the capacity to produce, release, and respond to acetylcholine (ACh), but the functional role of cholinergic systems in adult mammalian peripheral sensory nerves has not been established. Here, we have reported that neurite outgrowth from adult sensory neurons that were maintained under subsaturating neurotrophic factor conditions operates under cholinergic constraint that is mediated by muscarinic receptor–dependent regulation of mitochondrial function via AMPK. Sensory neurons from mice lacking the muscarinic ACh type 1 receptor (M1R) exhibited enhanced neurite outgrowth, confirming the role of M1R in tonic suppression of axonal plasticity. M1R-deficient mice made diabetic with streptozotocin were protected from physiological and structural indices of sensory neuropathy. Pharmacological blockade of M1R using specific or selective antagonists, pirenzepine, VU0255035, or muscarinic toxin 7 (MT7) activated AMPK and overcame diabetes-induced mitochondrial dysfunction in vitro and in vivo. These antimuscarinic drugs prevented or reversed indices of peripheral neuropathy, such as depletion of sensory nerve terminals, thermal hypoalgesia, and nerve conduction slowing in diverse rodent models of diabetes. Pirenzepine and MT7 also prevented peripheral neuropathy induced by the chemotherapeutic agents dichloroacetate and paclitaxel or HIV envelope protein gp120. As a variety of antimuscarinic drugs are approved for clinical use against other conditions, prompt translation of this therapeutic approach to clinical trials is feasible.
Authors
Nigel A. Calcutt, Darrell R. Smith, Katie Frizzi, Mohammad Golam Sabbir, Subir K. Roy Chowdhury, Teresa Mixcoatl-Zecuatl, Ali Saleh, Nabeel Muttalib, Randy Van der Ploeg, Joseline Ochoa, Allison Gopaul, Lori Tessler, Jürgen Wess, Corinne G. Jolivalt, Paul Fernyhough

Abstract
The current frontline symptomatic treatment for Alzheimer’s disease (AD) is whole-body upregulation of cholinergic transmission via inhibition of acetylcholinesterase. This approach leads to profound dose-related adverse effects. An alternative strategy is to selectively target muscarinic acetylcholine receptors, particularly the M1 muscarinic acetylcholine receptor (M1 mAChR), which was previously shown to have procognitive activity. However, developing M1 mAChR–selective orthosteric ligands has proven challenging. Here, we have shown that mouse prion disease shows many of the hallmarks of human AD, including progressive terminal neurodegeneration and memory deficits due to a disruption of hippocampal cholinergic innervation. The fact that we also show that muscarinic signaling is maintained in both AD and mouse prion disease points to the latter as an excellent model for testing the efficacy of muscarinic pharmacological entities. The memory deficits we observed in mouse prion disease were completely restored by treatment with benzyl quinolone carboxylic acid (BQCA) and benzoquinazoline-12 (BQZ-12), two highly selective positive allosteric modulators (PAMs) of M1 mAChRs. Furthermore, prolonged exposure to BQCA markedly extended the lifespan of diseased mice. Thus, enhancing hippocampal muscarinic signaling using M1 mAChR PAMs restored memory loss and slowed the progression of mouse prion disease, indicating that this ligand type may have clinical benefit in diseases showing defective cholinergic transmission, such as AD.
Authors
Sophie J. Bradley, Julie-Myrtille Bourgognon, Helen E. Sanger, Nicholas Verity, Adrian J. Mogg, David J. White, Adrian J. Butcher, Julie A. Moreno, Colin Molloy, Timothy Macedo-Hatch, Jennifer M. Edwards, Jurgen Wess, Robert Pawlak, David J. Read, Patrick M. Sexton, Lisa M. Broad, Joern R. Steinert, Giovanna R. Mallucci, Arthur Christopoulos, Christian C. Felder, Andrew B. Tobin

Abstract
Stroke is one of the most common diseases and a leading cause of death and disability. Cessation of cerebral blood flow (CBF) leads to cell death in the infarct core, but tissue surrounding the core has the potential to recover if local reductions in CBF are restored. In these areas, detrimental peri-infarct depolarizations (PIDs) contribute to secondary infarct growth and negatively affect stroke outcome. However, the cellular pathways underlying PIDs have remained unclear. Here, we have used in vivo multiphoton microscopy, laser speckle imaging of CBF, and electrophysiological recordings in a mouse model of focal ischemia to demonstrate that PIDs are associated with a strong increase of intracellular calcium in astrocytes and neurons. We found that astroglial calcium elevations during PIDs are mediated by inositol triphosphate receptor type 2–dependent (IP3R2-dependent) release from internal stores. Importantly,
Authors
Cordula Rakers, Gabor C. Petzold

Abstract
Intracerebral hemorrhage (ICH) is a devastating form of stroke that results from the rupture of a blood vessel in the brain, leading to a mass of blood within the brain parenchyma. The injury causes a rapid inflammatory reaction that includes activation of the tissue-resident microglia and recruitment of blood-derived macrophages and other leukocytes. In this work, we investigated the specific responses of microglia following ICH with the aim of identifying pathways that may aid in recovery after brain injury. We used longitudinal transcriptional profiling of microglia in a murine model to determine the phenotype of microglia during the acute and resolution phases of ICH in vivo and found increases in TGF-β1 pathway activation during the resolution phase. We then confirmed that TGF-β1 treatment modulated inflammatory profiles of microglia in vitro. Moreover, TGF-β1 treatment following ICH decreased microglial
Authors
Roslyn A. Taylor, Che-Feng Chang, Brittany A. Goods, Matthew D. Hammond, Brian Mac Grory, Youxi Ai, Arthur F. Steinschneider, Stephen C. Renfroe, Michael H. Askenase, Louise D. McCullough, Scott E. Kasner, Michael T. Mullen, David A. Hafler, J. Christopher Love, Lauren H. Sansing

Abstract
Hypertension is a leading risk factor for dementia, but the mechanisms underlying its damaging effects on the brain are poorly understood. Due to a lack of energy reserves, the brain relies on continuous delivery of blood flow to its active regions in accordance with their dynamic metabolic needs. Hypertension disrupts these vital regulatory mechanisms, leading to the neuronal dysfunction and damage underlying cognitive impairment. Elucidating the cellular bases of these impairments is essential for developing new therapies. Perivascular macrophages (PVMs) represent a distinct population of resident brain macrophages that serves key homeostatic roles but also has the potential to generate large amounts of reactive oxygen species (ROS). Here, we report that PVMs are critical in driving the alterations in neurovascular regulation and attendant cognitive impairment in mouse models of hypertension. This effect was mediated by an increase in blood-brain barrier permeability that allowed angiotensin II to enter the perivascular space and activate angiotensin type 1 receptors in PVMs, leading to production of ROS through the superoxide-producing enzyme NOX2. These findings unveil a pathogenic role of PVMs in the neurovascular and cognitive dysfunction associated with hypertension and identify these cells as a putative therapeutic target for diseases associated with cerebrovascular oxidative stress.
Authors
Giuseppe Faraco, Yukio Sugiyama, Diane Lane, Lidia Garcia Bonilla, Haejoo Chang, Monica M. Santisteban, Gianfranco Racchumi, Michelle Murphy, Nico Van Rooijen, Joseph Anrather, Costantino Iadecola










Copyright © 2024 American Society for Clinical Investigation
ISSN: 0021-9738 (print), 1558-8238 (online)